LQTS
Content is incomplete and may be incorrect. |
The Long QT Syndrome (LQTS) refers to a condition in which there is an abnormally long QT interval on the ECG. This was first recognized by Dr. Jervell and Dr. Lange-Nielsen in 1957. They described 4 children with a long QT interval which was accompanied by hearing deficits, sudden cardiac death and an autosomal recessive inheritance. [1]
The LQTS may be divided into two distinct forms: congenital LQTS and acquired LQTS. These forms may however overlap when QT prolongation due to medication occurs in a patient with congenital LQTS.
Diagnosis
General
- The diagnosis is by measurement of the heart rate corrected QT interval on the ECG, which can be calculated with the QTc calculator.
- Sometimes the QT interval can be difficult to assess. Read the guidelines for measurement of difficult QT interval.
- A QTc of > 500ms in patients with Long QT Syndrome is associated with an increased risk of torsade de pointes and sudden death. [2]
- In patients suspected of LQTS (e.g. family members of known LQTS patients) a QTc > 430ms makes it likely that a LQTS gene defect is present.[3]
- Because the QTc can change with age, it is best to take the ECG with the longest QTc interval for risk stratification. [4]
Physical examination
| Patients can present with symptoms of arrhythmias: |
|---|
|
Acquired LQTS
Acquired LQTS is most often caused by drugs that prolong the QT interval; combined with risk factors the risk of Torsade de Pointes is likely to increase.
Notorious QT prolonging drugs:[5] |
|---|
|
Concomittant risk factors for medication induced torsade de pointes: |
|---|
|
Congenital LQTS
The prevalence of congenital LQTS is about 1:3000-5000. More than 10 different types of congenital LQTS have been described. However, only LQTS 1-3 are relatively common. [6]
The three most common forms of LQTS can be recognized by the characteristic clinical features and ECG abnormalities.
| LQTS type | LQTS 1 | LQTS 2 | LQTS 3 |
|---|---|---|---|
| Gene/current | KCNQ1/IKs | KCNH2/IKr | SCN5A/INa |
| B-blokker efficacy | ++++ | + | ± |
| ECG | Early onset
broad based T wave |
Small late T wave | Late onset
T wave with normal configuration |
| Arrhythmogenic triggers | Exercise, especially swimming | Adrenergic triggers, especially nightly noise | Rest |
| Number of mutation carriers with events at age <40 | 25% | 50% | 50% |
| SCD incidence | 0,30% / year | 0,60% / year | 0,56% / year |
| Eponyme | If condition is homozygous:
Jervell and Lange-Nielsen syndrome 1 |
If condition is homozygous:
Jervell and Lange-Nielsen syndrome 2 |
Before the genes involved were known, some syndromes associated with a prolonged QT interval on the ECG had been described earlier:
- Anton Jervell and Fred Lange-Nielsen [1] from Oslo described in 1957 an autosomaal recessive syndrome that was associated with QT interval prolongation, deafness and sudden death: the now called Jervell-Lange-Nielsen syndrome.
- Romano-Ward syndrome is a long QT syndrome with normal auditory function and autosomal dominant inheritance.
- Andersen-Tawil syndrome was described in 1994 by Tawil et al. and was associated with potassium-sensitive periodic paralysis, ventricular ectopy and dysmorphic features (short stature, low-set ears, hypoplastic mandible, clinodactyly and scoliosis). It later appeared to be associated with a mutation in the KCNJ2 gene (LQTS type 7).
- Timothy syndrome is a LQTS syndrome (with frequently alternating T-waves) with webbing of fingers and toes, congenital heart disease, immune deficiency, intermittent hypoglycaemia, cognitive abnormalities and autism. It appeared to be caused by mutations in the CACNA1C gene (LQTS type 8).
Clinical diagnosis
Diagnosis of LQTS is established by prolongation of the QTc interval in the absence of specific conditions known to lengthen it (for example QT-prolonging drugs) and/or molecular genetic testing of genes associated with LQTS.
| Diagnostic criteria by Schwartz et al. | |||
|---|---|---|---|
| Findings | Points | ||
| ECG | QTc: | >480 ms
460-479 450-459 >480 during X-ECG |
3
2 1 1 |
| Torsade de pointes
T-wave alternans Notched T-wave in 3 leads Low heart rate for age (<2nd percentile for age) |
2
1 1 0.5 | ||
| Syncope: | With stress
Without stress |
2
1 | |
| Clinical history | Congenital deafness
Family members with definite LQTS |
0.5
1 | |
| Family history | Unexplained sudden cardiac death before age 30 | 0.5 | |
| Total score =1 Low probability; 1.5-3 Intermediate probability; =3.5 High probability | |||

The prolonged QT interval can cause torsade de pointes, which is usually self-terminating, thus causing a cardiac syncopal event. The mean age of onset of symptoms (syncope or sudden death) is 12 years and earlier onset is usually associated with more severe form of the disease. In LQTS type 1, cardiac symptoms are often precipitated by exercise; especially swimming is notorious for life-threatening cardiac events. In LQTS type 2, arrhythmogenic triggers are adrenergic; especially nightly noise (such as the morning alarm clock or nightly thunderlightening) is known to cause life-threatening cardiac events. On the other hand, in LQTS type 3, QT prolongation and possibly subsequent torsade de pointes is precipitated by bradycardia.
ECG tests

ECGs can be difficult because there is a considerable overlap between the QT interval of affected and unaffected individuals.
- The resting ECG is neither completely sensitive nor specific for the diagnosis of LQTS. The diagnostic criteria for the resting ECG are shown in the list of diagnostic criteria by Schwartz. Besides a prolonged QTc, the T-wave can have different patterns among the different genotypes.
- Holter recordings appear to be of minimal clinical utility from a diagnostic and prognostic prospective in evaluating LQTS
- The exercise ECG (X-ECG) commonly shows failure of the QT to shorten normally, thereby prolonging the corrected QT interval, and many individuals develop characteristic T-wave abnormalities.
- A brisk-standing test ECG, where the QT-interval is measured after abrupt standing with subsequent heart rate acceleration. There appears to be a form of QT-stretching and QT-stunning as demonstrated by Viskin et al. and Adler et al.
- Epinephrine infusion is a provocative test that increases the sensitivity of the ECG findings
- Adenosine infusion is a test provoking transient bradycardia followed by sinus tachycardia and therefore triggers QT changes that can distinguish patients with LQTS from healthy controls

A 12 lead ECG of a patient with genetically proven LQTS1

A 12 lead ECG of a patient with genetically proven LQTS2
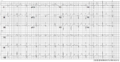
A 12 lead ECG of a patient with genetically proven LQTS3



{kind=link}
Genetic diagnosis
Today, 13 LQTS genes associated with LQTS have been identified. Most commonly, KCNQ1, KCNH2 and SCN5A, which are associated with LQTS type 1, type 2 and type 3 respectively, are found. Other, less frequently involved genes are displayed the table below.
| LQTS type | Gene | Protein (Ionchannel) | OMIM-link |
|---|---|---|---|
| Type 1 | KCNQ1 | KVLQT1 (IKs) | 607542 |
| Type 2 | KCNH2 | HERG (IKr) | 613688 |
| Type 3 | SCN5A | Sodium channel (INa) | 603830 |
| Type 4 | ANK2 | Ankyrin B (INa, K) | 600919 |
| Type 5 | KCNE1 | minK (IKs) | 176261 |
| Type 6 | KCNE2 | MiRP1 (IKr) | 613693 |
| Type 7 | KCNJ2 | Kir 2.1 (IK1) | 170390 |
| Type 8 | CACNA1C | Cav1.2 (ICa-L) | 601005 |
| Type 9 | CAV3 | Caveolin-3 | 601253 |
| Type 10 | SCN4B | Sodium channel (INa) | 608256 |
| Type 11 | AKAP9 | A-Kinase anchor 9 (IKs) | 604001 |
| Type 12 | SNTA1 | Syntrophin (INa) | 601017 |
| Type 13 | KCNJ5 | Kir3.4 (IK) | 600734 |
There is an important genotype-phenotype relationship on severity of the disease. In genotype–phenotype studies in the Rochester LQTS registry it was shown that in both LQTS type 1 and type 2, mutation locations and the degree of ion channel dysfunction caused by the mutations are important independent risk factors influencing the clinical course of this disorder.
Risk Stratification
Gene-specific differences of the natural history of LQTS have also been demonstrated and allow genotype-based risk stratification. Indeed, QT interval duration, gender and genotype (including mutation location and degree of ion channel dysfunction) are significantly associated with the outcome, with a QTc interval >500ms, and a LQT2 or LQT3 genotype determining the worst prognosis. Gender differently modulates the outcome according to the underlying genetic defect: the LQT3 males and LQT2 females are the highest risk subgroups. Risk stratification is best done by an expert cardio-genetics cardiologist.
Treatment [6]
Lifestyle modification
- No competitive sports in all LQTS patients
- Avoid QT-prolonging drugs in all LQTS patients
- No swimming in LQT1 patients
- Avoid nightly noise in LQT2 patients (e.g. no alarm clock)
Medication/Other therapies
- Beta-blockers are the cornerstone of therapy in LQTS. Beta-blockers even reduce the risk of sudden death in patients in whom a genetic defect has been found, but no QT prolongation is visible on the ECG and probably even in LQTS3 patients with bradycardia-associated cardiac events. In symptomatic patients propranolol and nadolol are equally effective, but a recent retrospective study showed that metoprolol should not be used for symptomatic LQT1 and LQT2 patients.[7]
- ICD implantation in combination with beta-blockers in LQTS patients with previous cardiac arrest, cardiac syncope or ventricular tachycardia while on beta-blockers. Symptomatic patients with LQTS type 3 can only be treated with an ICD with pacing possibilities, since their arrhythmic episodes are bradycardia-associated.
- Cardiac sympathetic denervation (LCSD) should be considered in the setting of beta-blocker breakthroughs, intolerance to pharmacotherapy and history of appropriate ICD therapies. Surgically, LCSD involves the resection of the lower half of the left stellate ganglion and the left-sided sympathetic chain at the level of T2, T3 and T4.
References
- JERVELL A and LANGE-NIELSEN F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957 Jul;54(1):59-68. DOI:10.1016/0002-8703(57)90079-0 |
- Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, Vicentini A, Spazzolini C, Nastoli J, Bottelli G, Folli R, and Cappelletti D. Risk stratification in the long-QT syndrome. N Engl J Med. 2003 May 8;348(19):1866-74. DOI:10.1056/NEJMoa022147 |
- Hofman N, Wilde AA, Kääb S, van Langen IM, Tanck MW, Mannens MM, Hinterseer M, Beckmann BM, and Tan HL. Diagnostic criteria for congenital long QT syndrome in the era of molecular genetics: do we need a scoring system?. Eur Heart J. 2007 Mar;28(5):575-80. DOI:10.1093/eurheartj/ehl355 |
- Goldenberg I, Mathew J, Moss AJ, McNitt S, Peterson DR, Zareba W, Benhorin J, Zhang L, Vincent GM, Andrews ML, Robinson JL, and Morray B. Corrected QT variability in serial electrocardiograms in long QT syndrome: the importance of the maximum corrected QT for risk stratification. J Am Coll Cardiol. 2006 Sep 5;48(5):1047-52. DOI:10.1016/j.jacc.2006.06.033 |
- Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004 Mar 4;350(10):1013-22. DOI:10.1056/NEJMra032426 |
- Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, Gregoratos G, Klein G, Moss AJ, Myerburg RJ, Priori SG, Quinones MA, Roden DM, Silka MJ, Tracy C, Smith SC Jr, Jacobs AK, Adams CD, Antman EM, Anderson JL, Hunt SA, Halperin JL, Nishimura R, Ornato JP, Page RL, Riegel B, Blanc JJ, Budaj A, Dean V, Deckers JW, Despres C, Dickstein K, Lekakis J, McGregor K, Metra M, Morais J, Osterspey A, Tamargo JL, Zamorano JL, American College of Cardiology/American Heart Association Task Force, European Society of Cardiology Committee for Practice Guidelines, European Heart Rhythm Association, and Heart Rhythm Society. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006 Sep 5;114(10):e385-484. DOI:10.1161/CIRCULATIONAHA.106.178233 |
- Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF, Hauer RN, Beckmann BM, Spazzolini C, Rordorf R, Rydberg A, Clur SA, Fischer M, van den Heuvel F, Kääb S, Blom NA, Ackerman MJ, Schwartz PJ, and Wilde AA. Not all beta-blockers are equal in the management of long QT syndrome types 1 and 2: higher recurrence of events under metoprolol. J Am Coll Cardiol. 2012 Nov 13;60(20):2092-9. DOI:10.1016/j.jacc.2012.07.046 |
- Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, and Bloise R. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001 Jan 2;103(1):89-95. DOI:10.1161/01.cir.103.1.89 |
- Shah M, Akar FG, and Tomaselli GF. Molecular basis of arrhythmias. Circulation. 2005 Oct 18;112(16):2517-29. DOI:10.1161/CIRCULATIONAHA.104.494476 |
- Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, Qi M, Vincent GM, Ackerman MJ, Kaufman ES, Hofman N, Seth R, Kamakura S, Miyamoto Y, Goldenberg I, Andrews ML, and McNitt S. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007 May 15;115(19):2481-9. DOI:10.1161/CIRCULATIONAHA.106.665406 |
- Roden DM. Clinical practice. Long-QT syndrome. N Engl J Med. 2008 Jan 10;358(2):169-76. DOI:10.1056/NEJMcp0706513 |
- Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Groffen AJ, Bikker H, and Christiaans I. Long QT Syndrome Overview. 1993.
-
Shimizu W, Moss AJ, Wilde AA, et al. Genotype-phenotype aspects of type 2 long QT syndrome. J Am Coll Cardiol 2009; 54:2052-62.
-
Sy RW, van der Werf C, Chattha IS, et al. Derivation and validation of a simple exercise-based algorithm for prediction of genetic testing in relatives of LQTS probands. Circulation 2011; 124:2187-94.
-
Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation 2011; 124:2181-4.
-
Viskin S, Postema PG, Bhuiyan ZA, et al. The response of the QT interval to the brief tachycardia provoked by standing: a bedside test for diagnosing long QT syndrome. J Am Coll Cardiol 2010; 55:1955-61.
-
Adler A, van der Werf C, Postema PG, et al. The phenomenon of "QT stunning": The abnormal QT prolongation provoked by standing persists even as the heart rate returns to normal in patients with long QT syndrome. Heart Rhythm 2012; 9:901-8.
-
Shimizu W, Noda T, Takaki H, et al. Diagnostic value of epinephrine test for genotyping LQT1, LQT2, and LQT3 forms of congenital long QT syndrome. Heart Rhythm 2004; 1:276-83.
-
Viskin S, Rosso R, Rogowski O, et al. Provocation of sudden heart rate oscillation with adenosine exposes abnormal QT responses in patients with long QT syndrome: a bedside test for diagnosing long QT syndrome. Eur Heart J 2006; 27:469-75.